Paper Walkthrough: P-Net - a biologically informed deep neural network for prostate cancer discovery
In which I cover a new biologically informed neural network architecture.

In this post I'll be covering a recent nature paper from Elmarakeby et al. [1] introducing a deep learning model for predicting clinical outcome in prostate cancer. The model uses molecular data (specifically mutation status and copy number, although other gene-level features could also be used) to model overall survival of patients with prostate cancer.
The novelty of this method lies in the use of a biologically informed model (more on what is meant by that later). This flavor of model has two distinct advantages over previous clinical deep learning models: 1) the model is simpler to interpret, and 2) the number of learnable parameters in the model is dramatically lower than for a traditional densely connected network with the same number of nodes.
A biologically informed network
In a vanilla densely connected neural network, each node in a layer is connected to every node in the subsequent layer. With P-net however, these connections are trimmed so only nodes with biological connection to each other are connected.
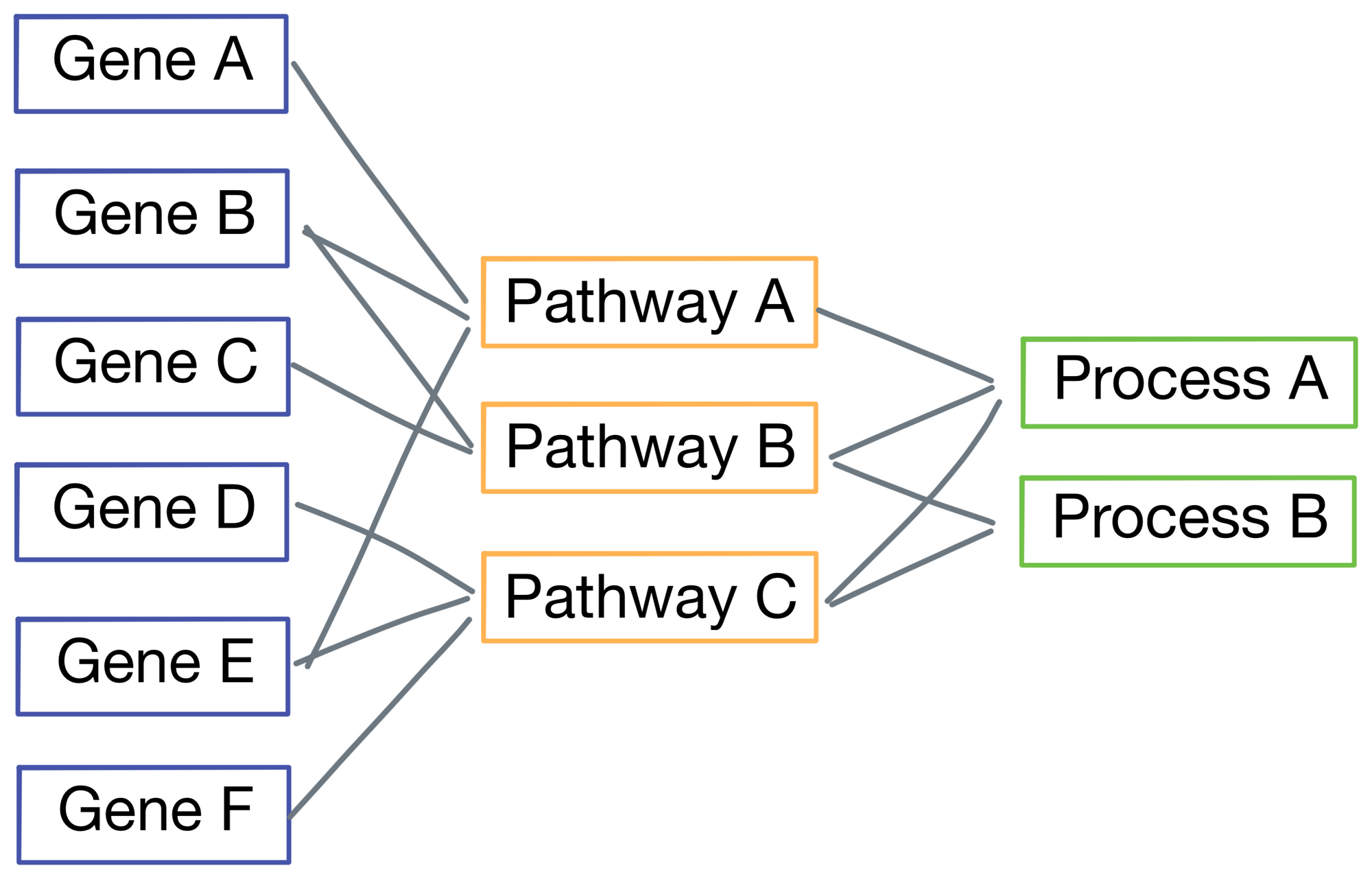
Specifically, P-net is hierarchical, meaning early layers in the network represent finer-grained biological phenomenon, while later layers represent increasingly macro-level biological pathways and features. Below is a simplified representation of P-net with just 3 layers (the version from the paper actually has 7, but using 3 here for visual simplicity). Connections between nodes are based on known biological phenomena. For example, in the figure Gene A and Gene B are part of Pathway A, while Gene C and D are members of Pathway B. The connections are obtained from the Reactome database.
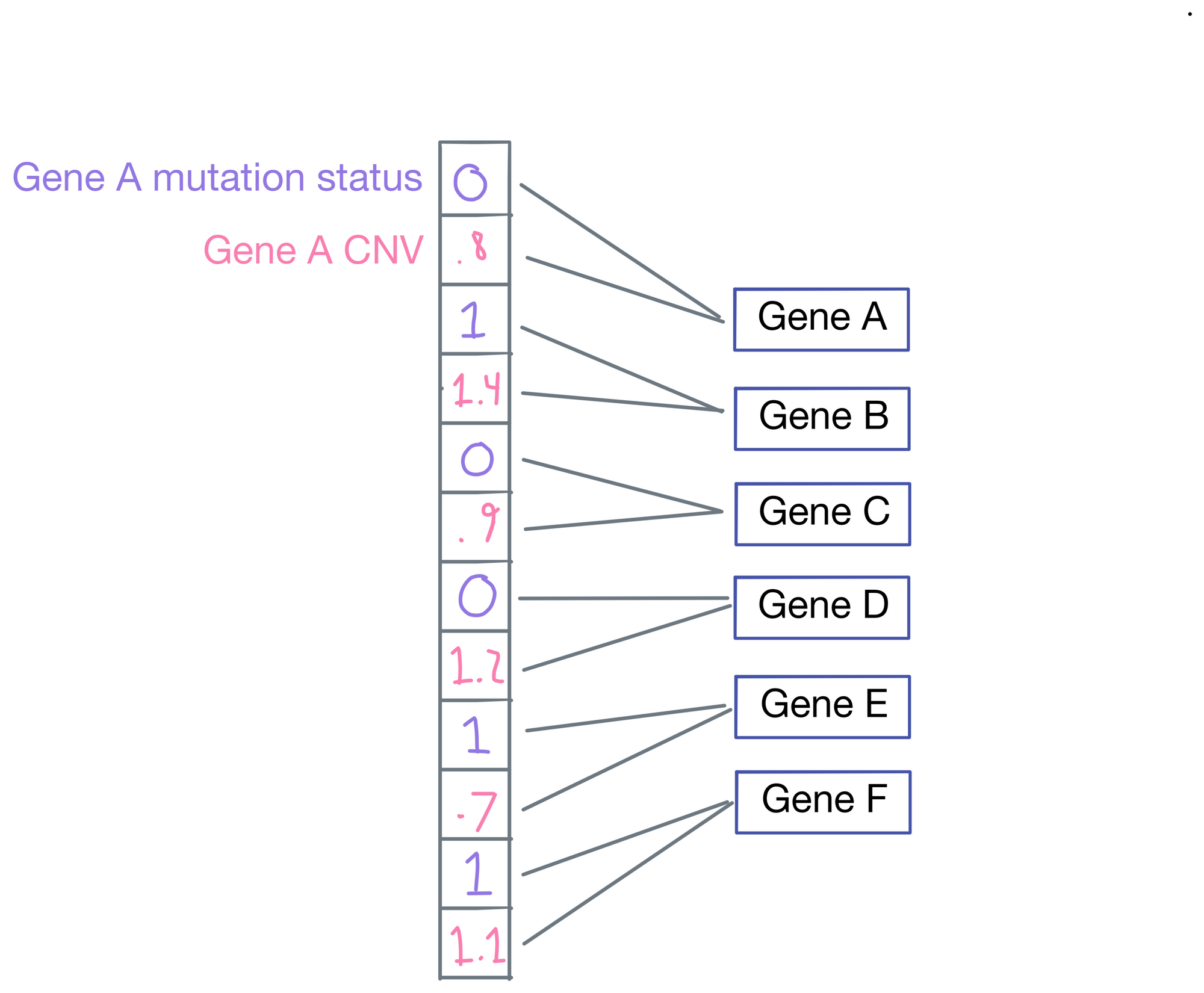
If we zoom in on the first layer, the network inputs are a vector representing mutation status (1 for mutated, 0 for wild type), and copy number (lower numbers are deletions and higher numbers are amplifications). Importantly, each input is only connected to its corresponding gene.
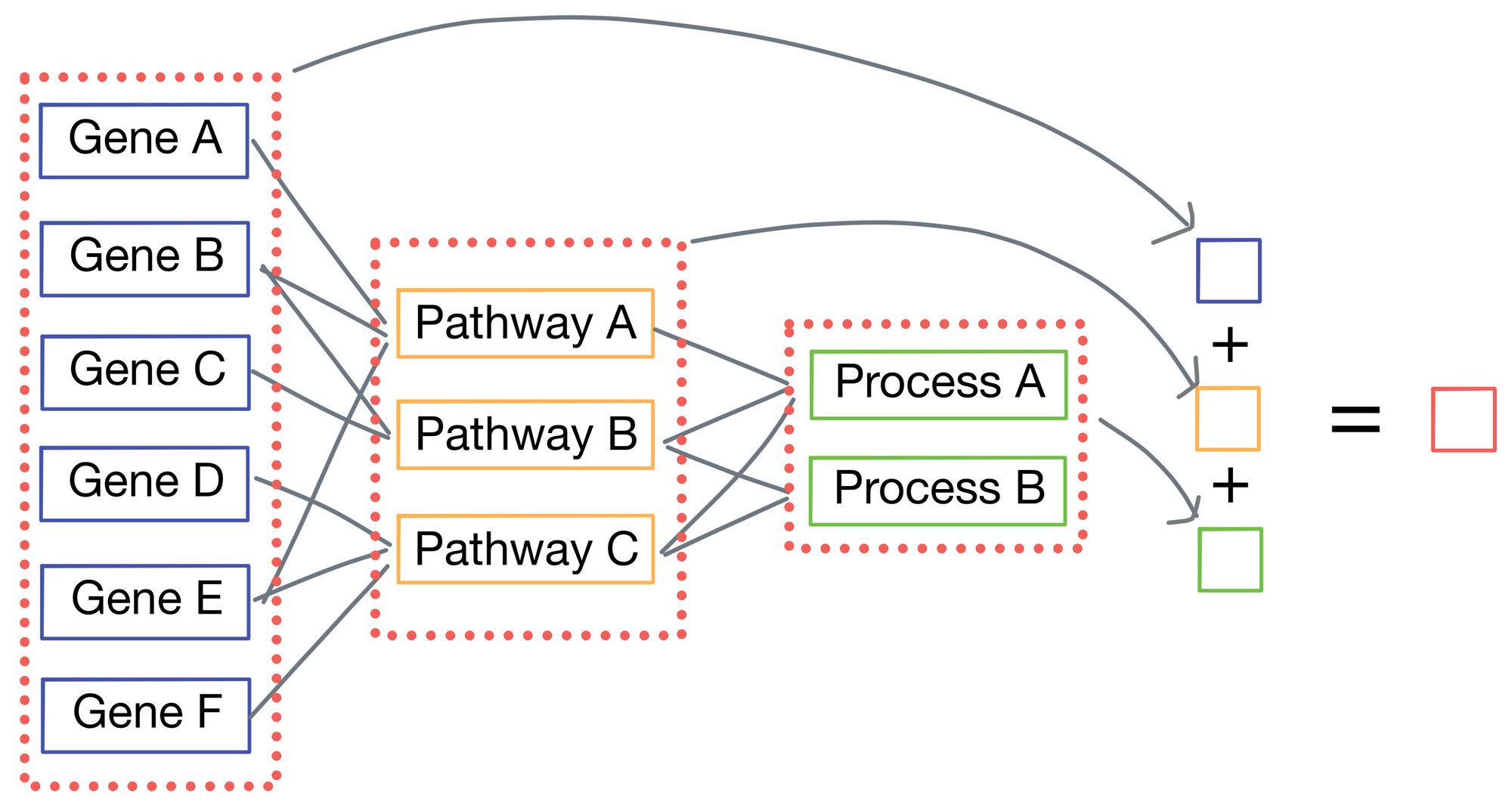
P-net is a regression model, as the ultimate goal of P-net is to predict patient survival. Typically, the last layer of the network would be connected to a length 1 vector representing survival time, but P-net takes a different approach. Instead, each layer produces its own output in addition to its connections to the subsequent layer. Each of these outputs are then averaged to each other to come up with a final survival prediction.
Implementation of skipped connections
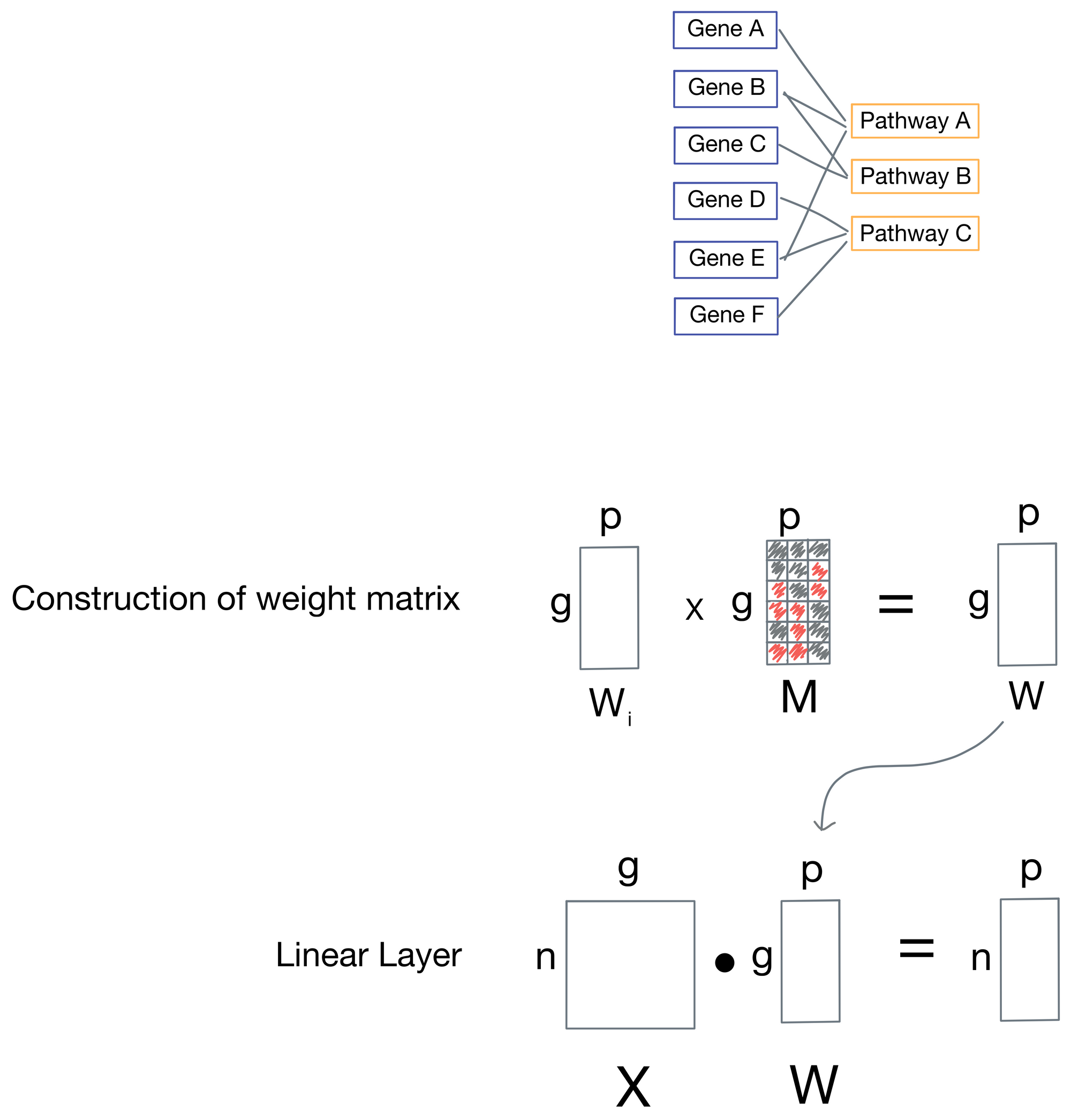
Now that we've enumerated the neural network architecture, how are the skipped connections implemented? It turns out to be fairly simple: for each weight matrix (one is associated with every layer), there is a corresponding mask matrix M that is used to set positions in the weight matrix representing connections to nodes that are not biologically relevant to zero.
Explainability
Another advantage of P-net is increased biological interpretability due to both it's lack of irrelevant connections and each layer having a direct connection to the network output. The authors use the DeepLift [2] algorithm to compute importance scores for each node in the network by tracing the connections from the output to each node in the network.
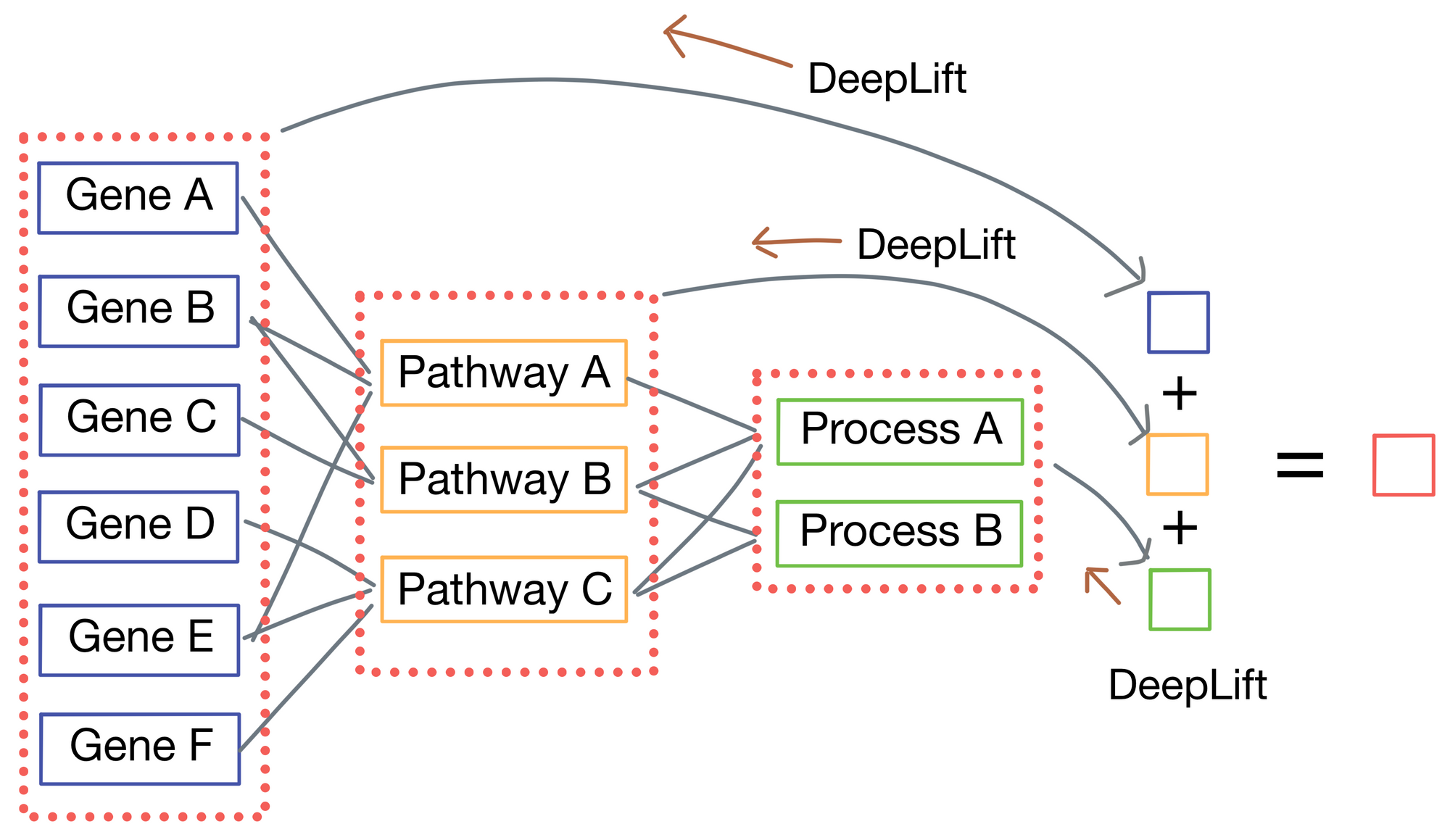
In doing so, the authors can cleanly see with genes and pathways most influence overall survival of patients in their sample cohort. When examining these importance scores they see numerous genes and pathways known to be important to prostate cancer biology such as TP53. However, they also discover two new genes, MDM4 and FGFR1, previously unknown to the field and confirmed their role in tumor progression by testing them in-vitro. Further, MDM4 is also targetable with therapeutics so it could constitute a novel target for treatment in prostate cancer.
I believe we will start to see a proliferation of these types of biologically informed neural networks because they are much more adept at determining genes and pathways related to whatever target the network is trying to predict.
Hope you enjoyed the post. Cheers :)
1. Elmarakeby et al.: https://www.nature.com/articles/s41586-021-03922-4
2. DeepLift: https://arxiv.org/abs/1704.02685