Enformer: Effective gene expression prediction from sequence by integrating long-range interactions
Paper walkthrough of the Enformer introduced in "Effective gene expression prediction from sequence by integrating long-range interactions" by Avsec et al.

Prediction of genomic elements from sequencing data is an open question in biology. Recent methods such as DeepC [1] (to predict chromatin architecture) and Basenji2 [2] (to predict expression and chromatin architecture) have been developed towards this end. However, results vary compared to experimental baselines, and more work is needed to close the gap between predictions and empirical results.
Enter Enformer [3].
The Enformer is inspired by Basenji2, the previous state-of-the-art for genomic track prediction from DNA sequences. Recently, the transformer architecture has been shown to well relate distal elements of a sequences. Enformer embraces this idea by co-opting several elements of the Basenji2 architecture, namely convolutional blocks. However, it replaces the dilated convolutions of Basenji2 with transformer layers. This swap allows Enformer to see a much wider range of the DNA sequence than Basenji2 (>100kb vs. < 20kb). Further, it allows the Enformer to more efficiently route information between distant regions of the sequence. This is crucially important because it is known biology that noncoding genomic regions far from actual genes themselves can have impact on the expression of those genes.
So what does the Enformer actually look like?
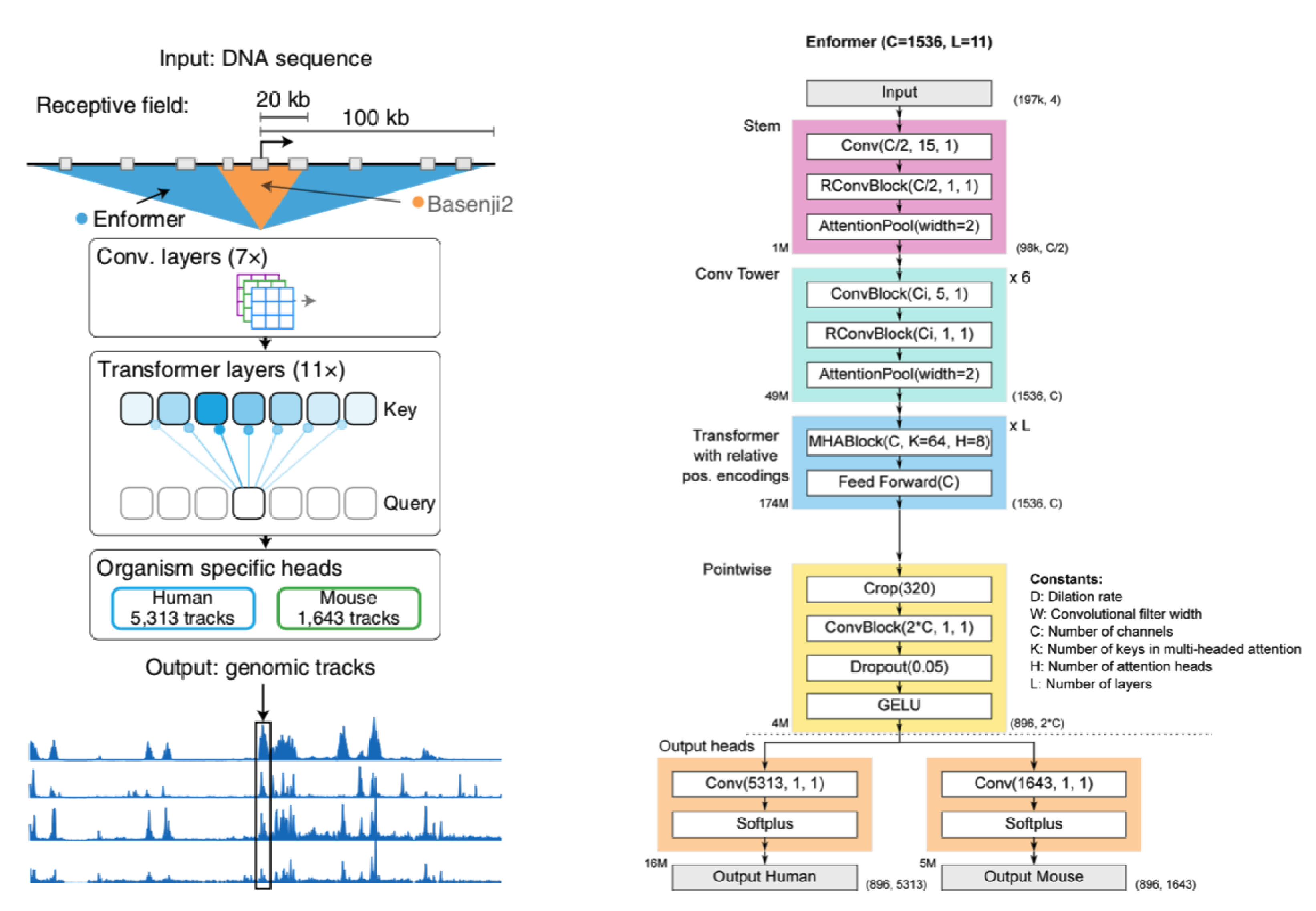
Before I walk through a generalized version of the Enformer, let's take a broad look at whats going on.
We start with a DNA sequence that is ~200kb (kila-bases) long. This sequence is pushed through a series of convolution layers. The purpose of said layers is to compress and summarize information in the sequence to a length that is suitable for transformer input. The transformers job is then to associate regions of DNA (which may be far away from each other) and route information between them. In other words, it gives the Enformer the ability to see the entire DNA sequence, where as Basenji2 could only see regions of ~20kb in size.
After the transformer layers the model spits out genomic information in the form of genomic tracks, which show gene expression and various chromatin features for sequential positions in the genome. Enformer predicts the following tracks: Cap Analysis Gene Expression (CAGE), Histone modification (ChiP Histone), TF binding (ChiP TF), and DNA accessibility (DNase/ATAC). Importantly, Enformer is trained on both human and mouse genomes by alternating batches of human and mouse inputs during training.
Before I get into a breakdown of Enformer and its components, a quick note: for ease of explanation, the architecture I describe is not an entirely faithful reconstruction of Enformer (for that see the detailed architecture in the panel above).
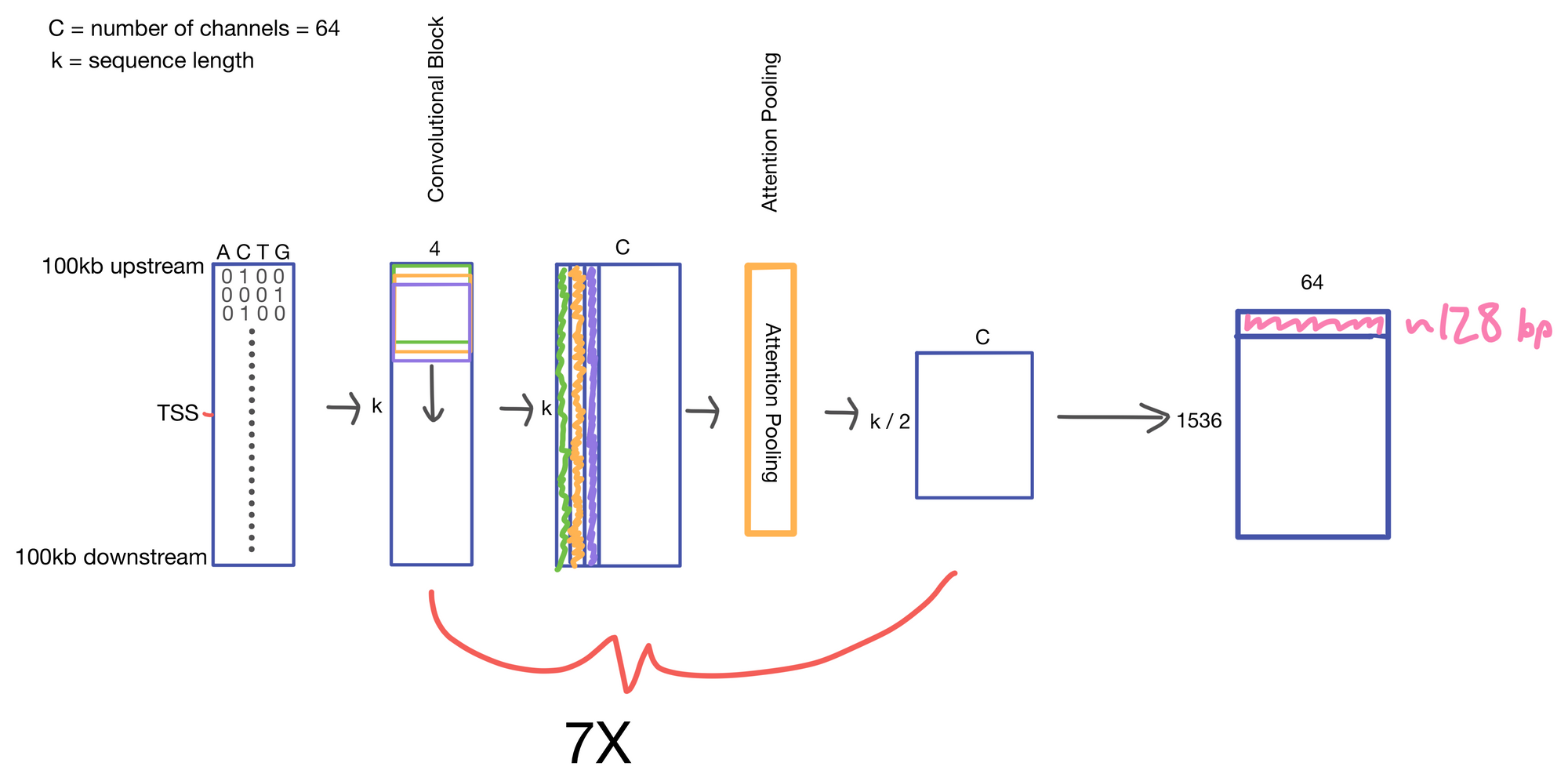
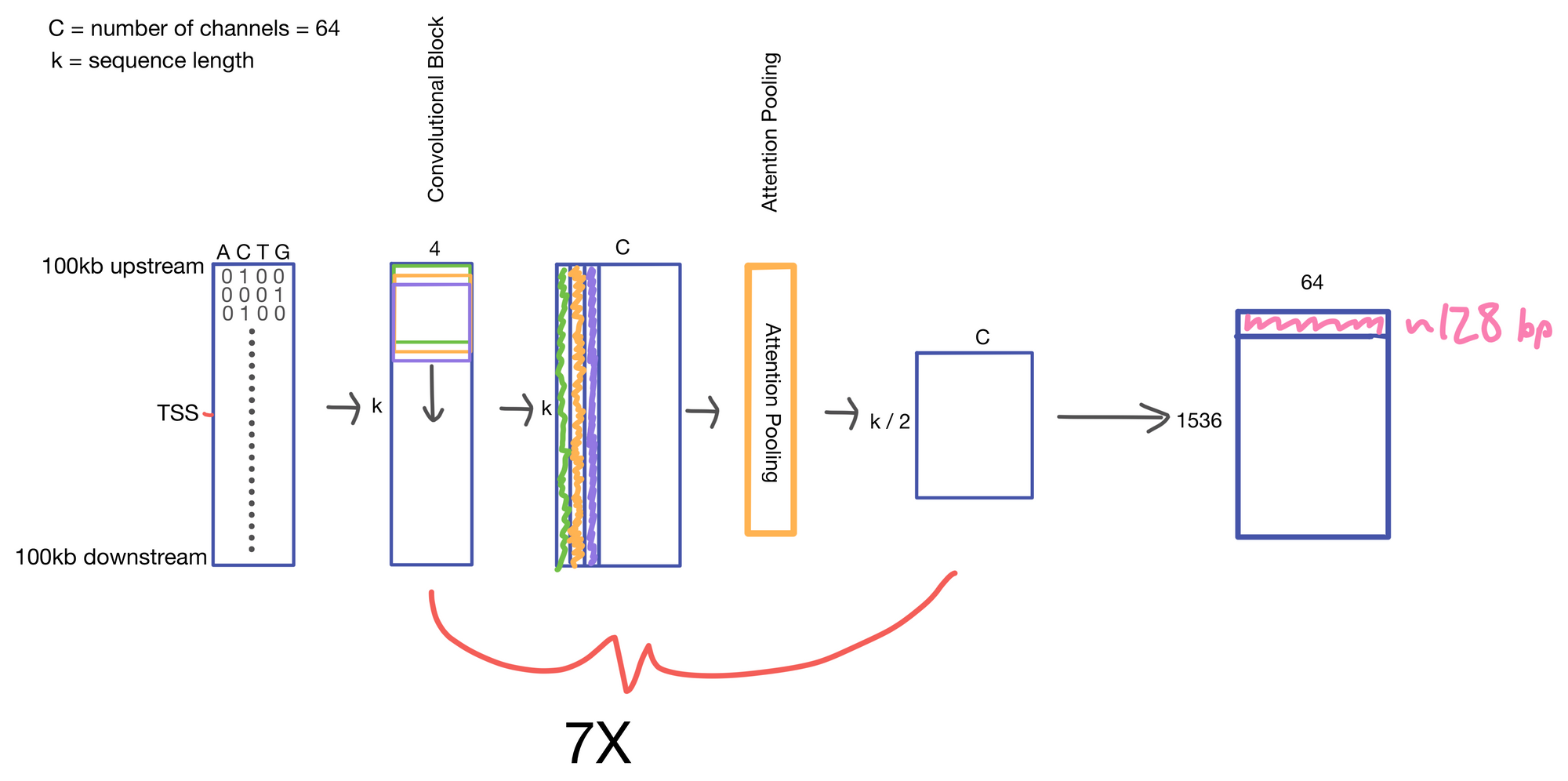
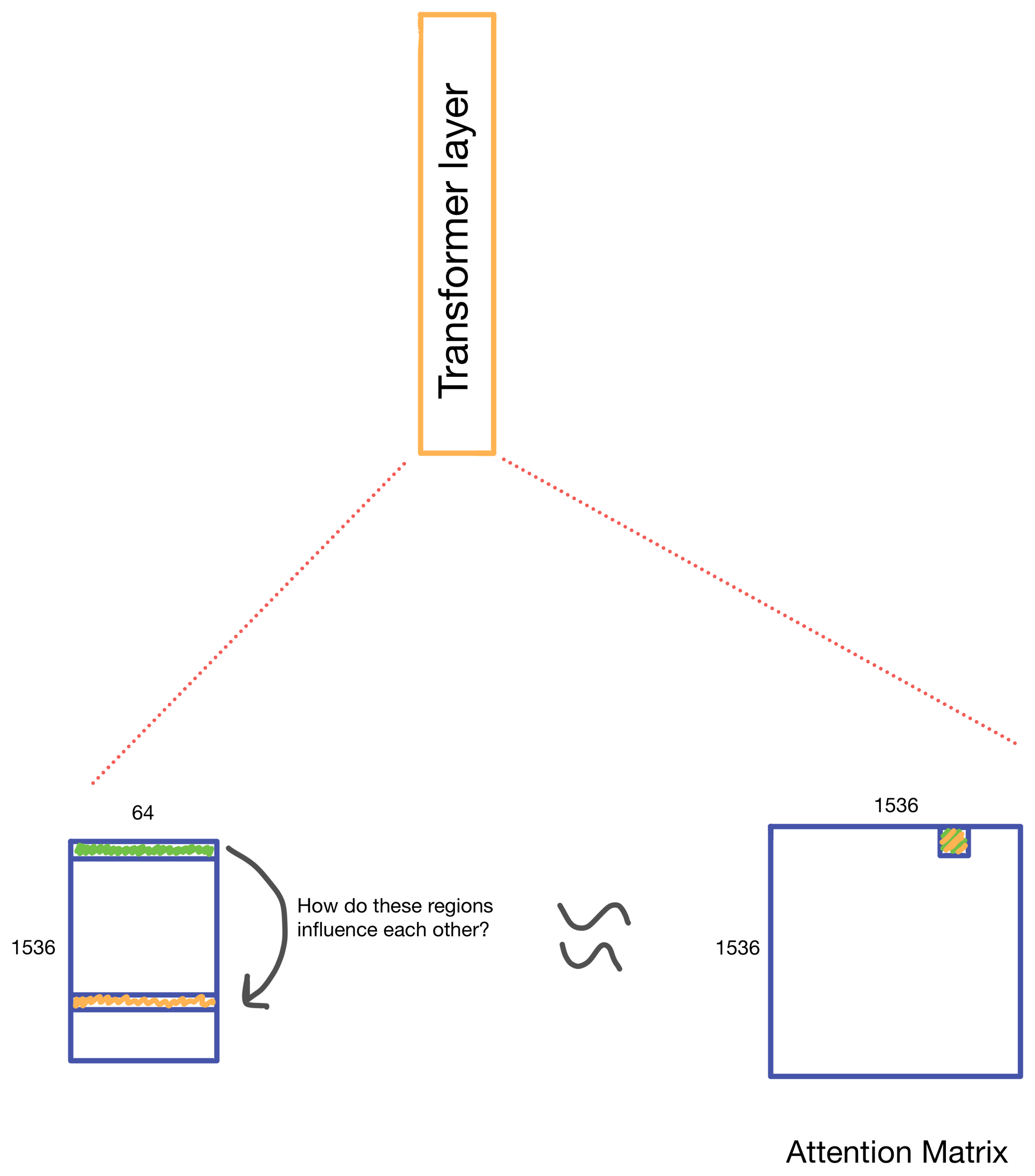
We start with a matrix representing our one hot encoded input DNA sequence that is centered at the transcription start site (TSS) of the gene whose genomic track we are interested in. A convolutional and attention pooling layer are then used to summarize and compress information in the DNA sequence. This step is repeated 7 times. Each time through the convolutional block, the sequence length is cut in half, giving us a final tensor shape of 1,536 x 64. A suitable matrix size for transformers to work. Of note, each row of the resulting matrix represents a region approximately 128 (27) bp long.
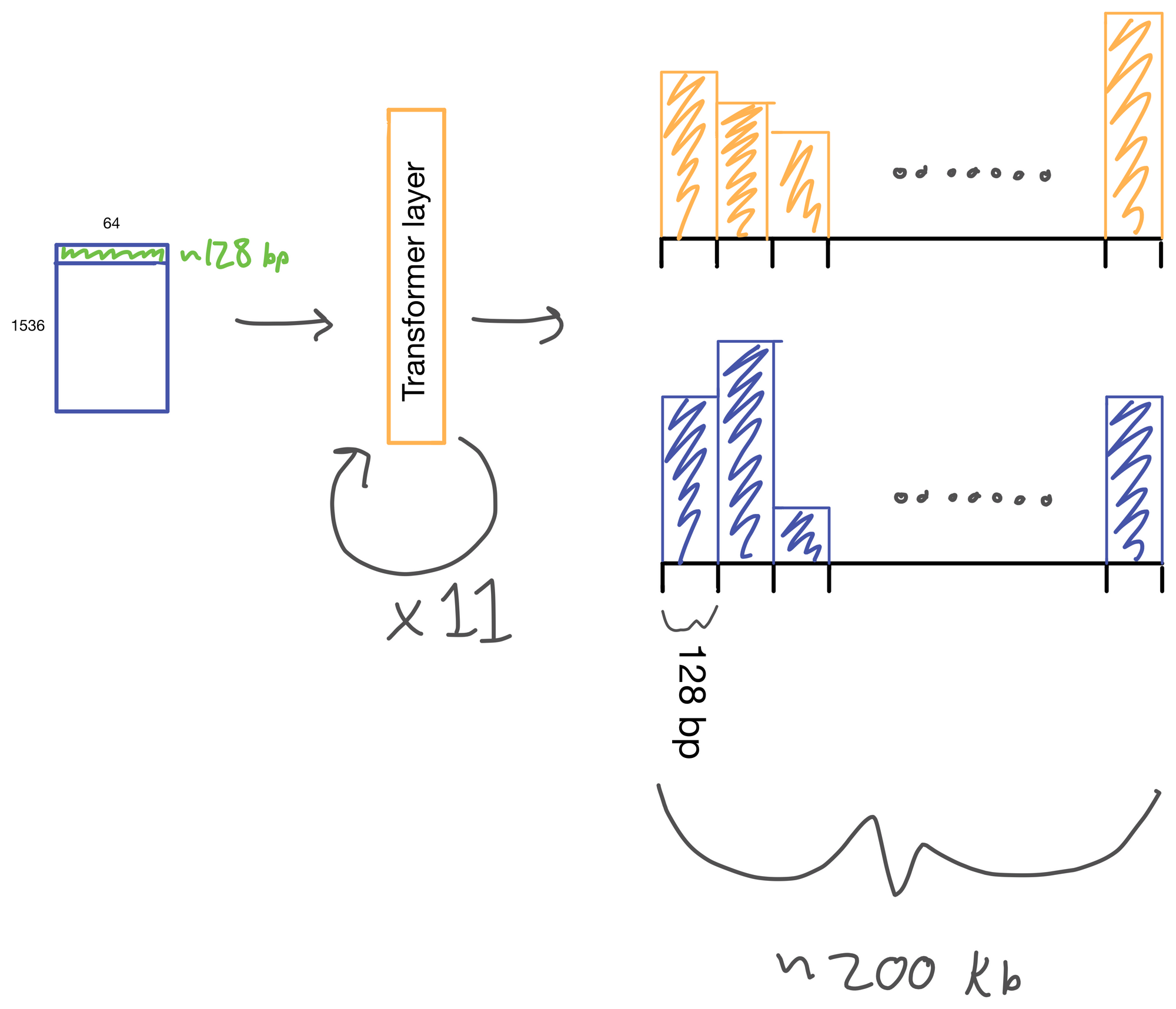
The suitably compressed matrix is then fed into a series of 11 multi-head attention layers. After which the Enformer predicts various genomic tracks (which have been aggregated into regions of 128bp).
I wont detail transformer layers here, but if you're interested see my two part post on multi-head attention here and here, which describes a transformer layer that is similar to the one used in Enformer. The transformer layer lives to relate regions of the DNA sequence input to on another. In Basenji2 this was accomplished with dilated convolutions. However, this approach both limits the receptive view of the layer and information from regions far away from each other can be diluted. Transformers don't have this issue. Signal is preserved from distant regions, and they excel at routing information between distal regions of sequences.
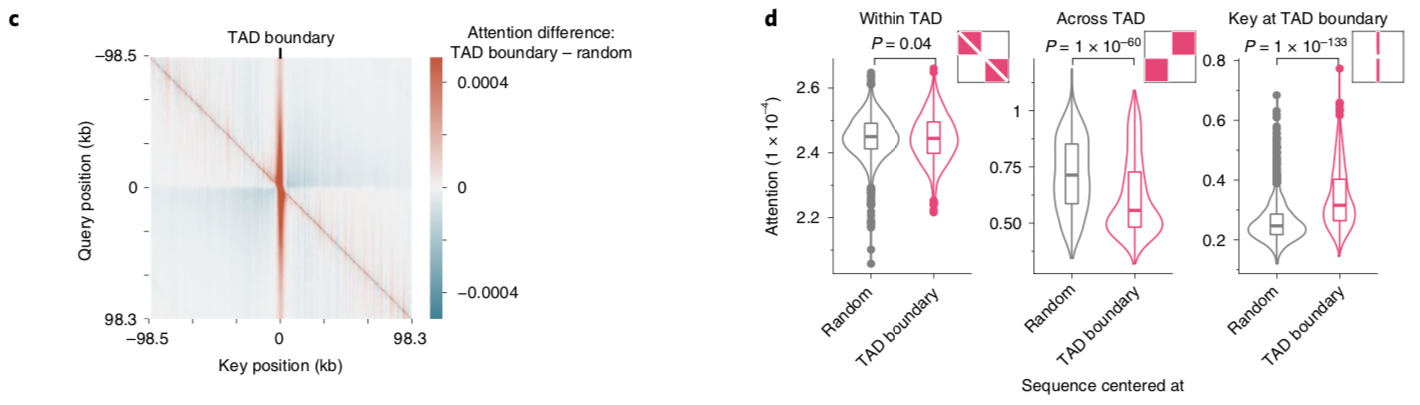
A nice byproduct of the transformer layers is the attention matrix . This matrix describes how the transformer routs information. Meaning it contains information on how much each region impacts every other region. This matrix is taken advantage of in the results section of the paper where the authors show that regions with the most attention are known promoters and topologically associated domains (TADs), which are known to influence gene expression.
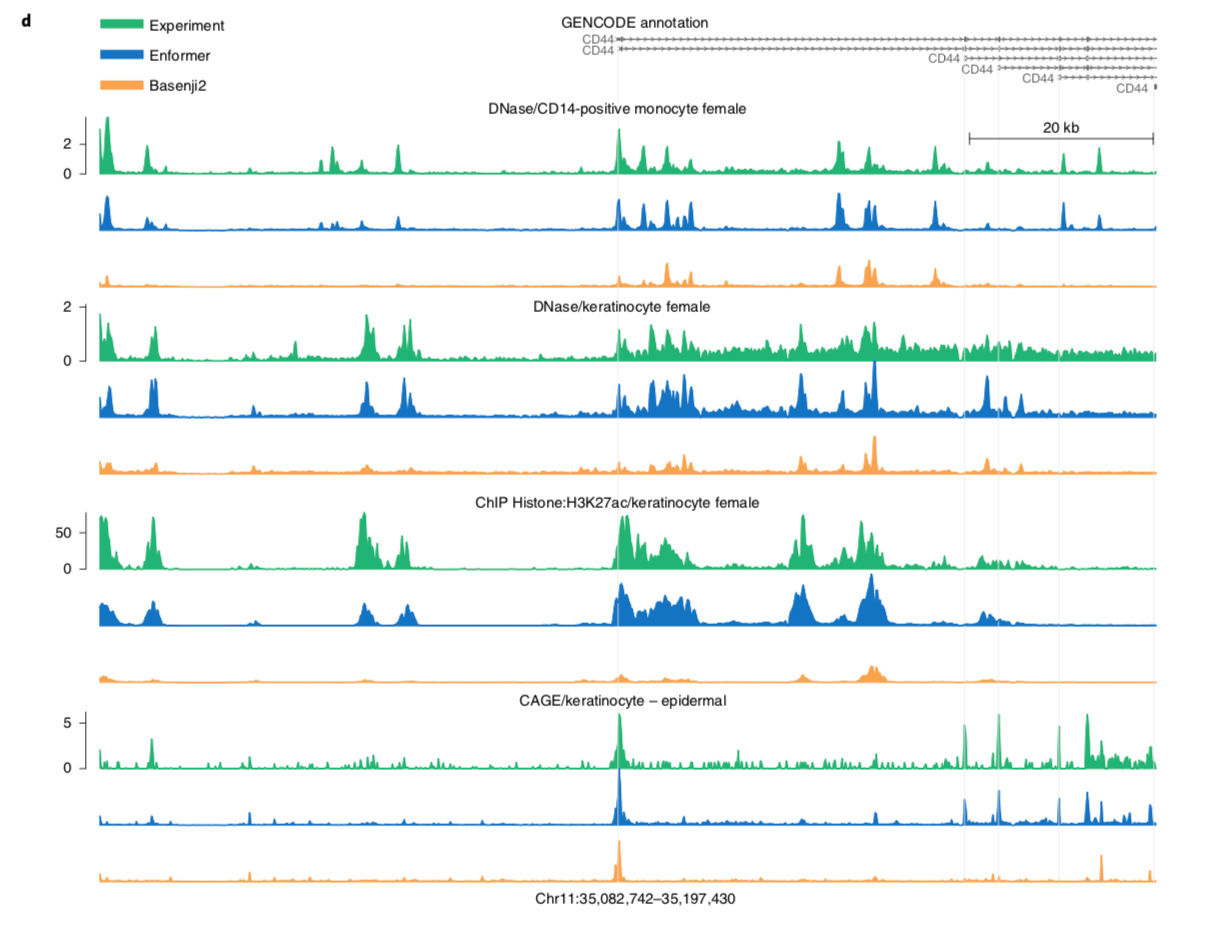
I'll leave you with a figure showing Enformer predictions vs. and experimental baseline.
Hope you enjoyed the post :)
1. DeepC: predicting 3D genome folding using megabase-scale transfer learning, https://www.nature.com/articles/s41592-020-0960-3
2. Sequential regulatory activity prediction across chromosomes with convolutional neural networks
, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5932613/
3. Effective gene expression prediction from sequence by integrating long-range interactions
, https://www.nature.com/articles/s41592-021-01252-x